
Essential Guide to Medical Electronic Device Design
What is Medical Electronic Device Design?
Medical electronic device design involves creating electronic systems that interact with the human body and must meet strict regulatory, safety, and performance standards (such as IEC-60601 and FDA requirements). These devices must pass EMI/EMC, ESD, and power immunity testing before reaching the market.
The medical electronic device industry grew significantly over the last few decades following the exponential advancements in electronics, computation, and life sciences. Also, the rise of personal and mobile healthcare increased the demand for small, wearable, and low-power technologies, adding heat to the medical devices market. Therefore, it is not surprising that more and more electronics developers are entering the field. But designing medical electronics is not an easy task, and the developer must be prepared to face challenges and regulations that are not required in consumer electronics.
One of the most common mistakes is not considering the relevant regulations early in the design process, leading to expensive modifications or even the complete redesign of the device. Therefore, it is important to know the regulations and best practices before starting a new project. In this article, we will discuss the most important regulations and standards for medical electronic devices, focusing on electronic systems.

Tests & Standards for Medical Electronics
What standards do medical electronic devices need to meet?
Medical devices must comply with IEC-60601 standards and pass safety, EMC, and immunity testing to ensure safe operation in real-world environments.
The regulation for medical devices depends on the country of origin:
| Region | Regulatory Body |
|---|---|
| United States | FDA |
| Europe | EMA |
Design guidelines are typically based on:
- IEC-60601-1
- IEC-60601-1-2
By adhering to these standards, the product becomes “601 compliant,” which is crucial for certification approval and market release.
The IEC-60601-1 defines medical electrical equipment as any device that transfers or detects energy to or from the patient.
Common examples:
- ECG, EEG, EMG sensors
- Ultrasound systems
- Heart rate monitors
- Glucometers
- Thermometers
- Pacemakers
These devices must pass EMI/EMC, ESD, and power supply immunity tests while maintaining safety and performance.
EMI/EMC Tests
What do EM/EMC tests ensure?
They ensure a device does not emit harmful interference and continues to operate correctly in electromagnetic environments.
Devices must comply with CISPR 11 for emissions.
Types of EMI
| Type | Description |
|---|---|
| Conducted EMI | Through cables |
| Radiated EMI | Wireless interference |
Frequency ranges (CISPR 11)
- Conducted: 150 kHz – 30 MHz
- Radiated: 150 kHz – 18 GHz
Maximum amplitudes depend on device class.
Conducted power harmonics must comply with IEC 61000-3-2, especially in switched-mode power supplies.
Common mitigation methods:
- Ferrite beads
- Line filters
- Chokes
Radiated EMI regulations vary by country.
Radiofrequency susceptibility is critical in medical devices. IEC 60601-1-2 defines immunity testing:
- 80 – 2700 MHz (radiated)
- 0.15 – 80 MHz (conducted)
Devices are tested in an anechoic chamber, and performance is monitored during and after exposure.
They must also withstand low-frequency magnetic interference (50/60 Hz) per IEC 61000-4-8.
Design considerations:
- Avoid high impedance nodes
- Avoid loops and parasitic antennas
- Use shielding and differential signaling
These techniques significantly improve EMI immunity.
Power Supply Immunity Tests
Why are power immunity tests important?
They ensure devices remain safe and functional despite voltage fluctuations or instability. Key standards:
| Standard | Purpose |
|---|---|
| IEC 61000-3-3 | Voltage fluctuation |
| IEC 61000-4-11 | Voltage dips |
| IEC 61000-4-4 | Burst testing |
| IEC 61000-4-5 | Surge testing |
Medical devices must operate during voltage dips of 95% for up to 5 seconds, typically handled with batteries or capacitor banks.
Burst and surge testing evaluates tolerance up to ±2 kV.
Electrostatic Discharge Tests
What is ESD testing?
It ensures devices can withstand electrostatic discharge without failure. Devices are tested under:
- ±8 kV (contact)
- ±15 kV (air)
This is especially critical for devices in direct contact with patients and high-voltage equipment like defibrillators.
Risk Management Matrix
What is a risk management matrix?
A tool (ISO 14971) used to evaluate risk based on likelihood and severity.
The international guidelines for the matrix are defined by the ISO 14971:2019 standard, which also includes medical software and in-vitro devices. It addresses risks related to biocompatibility, electrical harms, radiation, and usability among others.
A risk management matrix can be designed by distributing:
- Failure likelihood (rare, unlikely, likely, expected, and certain)
- Consequences (minor, moderate, serious, major, and catastrophic) and filling the cells with the level of acceptance.
One example is shown in Table 1.
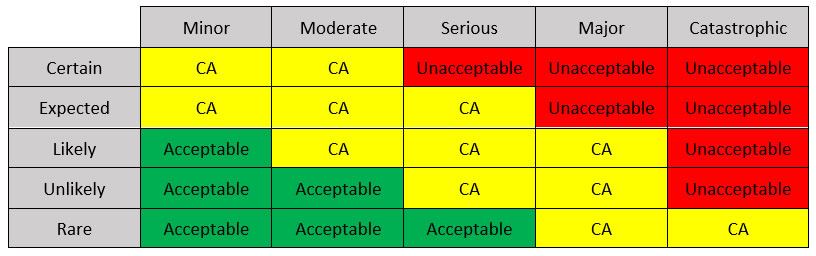
Table 1: A possible Risk Management Matrix (CA: conditionally acceptable).
Patient Isolation
Why is patient isolation critical?
It prevents electrical harm by ensuring no direct electrical path between device and patient.
Requirements include:
- No DC path to patient
- Controlled leakage current
- Proper insulation and clearance
Common isolation methods:
- Isolation transformers
- Optocouplers
- Capacitive coupling
All power systems must comply with IEC-60601-1.
Laboratory Equipment
Devices with no patient contact must comply with:
- IEC-61010
- IEC-60810-1
Examples:
- Microscopes
- Autoclaves
- Chemistry analyzers
- Incubators

Medical Electronics Certification Path
An integral part of the medical design loop is the certification process, which decides whether your device is ready to market or not. This process is defined, controlled, and executed by a regulatory agency, varying from country to country. For instance, medical devices are regulated by the FDA in the US, by the EMA in Europe, and Health Canada in Canada. In this article, we are going to focus on the FDA regulation, as the US medical market is huge, and the FDA complies with most regulations around the world.
FDA Device Classes & Market Release
The FDA rules for product certification depend on the nature of the medical device. The agency defines three different device classes to specify what type of process the product must undergo, including premarketing application, approval, and controls. Medical devices are classified according to their risk level:
| Class | Risk Level | Examples |
|---|---|---|
| Class I | Low | Bandages, masks |
| Class II | Moderate | X-rays, infusion pumps |
| Class III | High | Pacemakers |
The FDA defines several steps and standards to bring a medical device to the market, including general and specific controls, pre-market notification – 510(k), and premarket approval (PMA). The certification path depends on the class and nature of the device, as well as the exemptions applied. Therefore, the first step consists of identifying the class of the device.
1. Classification Panels
One of the earliest ways of identifying the class of a device is by finding a matching description in the classification panels. The FDA divided more than 1700 types of medical devices into 16 different panels (or categories).
- Each panel is associated with a medical specialty, including “General Hospital”, “Microbiology”, and “Orthopedic”, which are then divided into device types.
- For instance, the “Orthopedic” specialty covers more than a hundred types, such as “Bone Cap” and “Intramedullary Fixation Rod”.
- Each item contains a brief description and the corresponding class (I, II, or III) of the device. The classification panels can be accessed on the FDA website.
2. General and Special Controls
The FDA also establishes a basic set of design rules that are common to all medical devices: the general controls. It regulates, among other items, device adulteration, misbranding, registration, premarket notification, and restricted uses. It also provides special guidelines for manufacturing practices.
- Class I Devices: In the case of exempt Class I devices, compliance with the general controls is the only requirement before market release.
- Class II Devices (Special Controls): For non-exempt Class II devices and most Class II devices, the General Controls are not enough, so the FDA defines the Special Controls. Post-market surveillance, premarket data requirements, special labeling requirements, and patient registries are some of the topics included in the Special Controls section.
3. Pre-market Notification 510(k)
In addition to the Special Controls, Class II devices require pre-market notification submission for FDA approval. This process is often called 510(k), which corresponds to the premarket notification section in the Food, Drug, and Cosmetic Act (FD&C).
- The 510(k) submission is a thorough description of the medical device, including technical and functional information concerning effectiveness, safety, and operation modes.
- For approval, the FDA verifies the device is “substantially equivalent” to another medical device already in the market. Therefore, the 510(k) process is basically a certification by equivalency.
- Most Class III and some exempt Class II devices only need to submit a 510(k) notification before market release, so searching for equivalent equipment to avoid the need for a PMA is a fundamental step in these classes.
4. Pre-market Approval (PMA)
Finally, the last step in the FDA certification process is the Pre-Market Approval (PMA). Most Class III devices are considered risk equipment, and cannot be marketed legally without a PMA certification.
- The PMA process includes a series of clinical trials and laboratory experiments to certify the effectiveness and safety of the medical device through sound and consistent data. Therefore, it is an expensive, laborious, and time-consuming process, and fulfilling the PMA can be disastrous to the company.
- The FDA provides well-defined guidelines and standards for every test, which can be consulted by the engineer during the design process to prevent recalls. It is important to identify the class of the device and integrate the PMA requirements early on the design flow.
Coventry Wipes, Swabs & Solvents for Critical Medical Electronics
Coventry™, a brand of Chemtronics, is known globally for solving the most critical precision cleaning challenges. Coventry has a wide variety of cleanroom swabs and wipes to meet your most demanding applications. The swabs are produced using a proprietary foam process that prevents scratching of delicate surfaces. Also included in the product line are sealed fabric, sealed foam, and static control.
Coventry solvent cleaners and carrier fluids are formulated and manufactured to an exacting specification to provide unparalleled performance. We have off-the-shelf products and can even engineer custom solutions to meet your specific needs.
- Coventry ESD Static Control Swabs: Engineered to eliminate damage from static electricity. The handles of these swabs will dissipate 99% of a 5,000 volt (5 KV) charge in less than 2.0 seconds. The static dissipative plastic alloy used to make the handles is inherently static dissipative. It contains no additives, coatings, or surfactants so it functions at any level of humidity and is very resistant to solvents—even acetone. Choose between our Class 100 (ISO Class 4) laundered polyester or 100 pore-per-inch polyurethane swabs.
- Coventry 12320 Precision Cleaning Solvent: A proprietary azeotropic blend designed specifically for effective cleaning and validation of NASA Propulsion Oxygen Systems.
Hardware. This unique formulation is non-combustible and exhibits good material compatibility, oxygen system compatibility, and high cleaning effectiveness. Coventry 12320 is an ideal AK-225 replacement in systems where high cleaning efficiency and high non-volatile residue removal are required.
Coventry™ 12808 Multi-Phase Carrier Solvent: A saturated mixture of n-octane and perfluorocarbon (PFC). The n-octane provides solvency while the PFC provides an inert perfluorocarbon vapor blanket above the liquid mixture. These vapors act to impede the flammability of the n-octane. As the perfluorocarbon evaporates during use, the supplemental PFC moves into the saturated solution.
Chemtronics also offers a complete line of vapor degreasing and ultrasonic solvents that are ideally suited for critical medical electronics. For more information, contact your Chemtronics application specialist at 678-596-5534 or [email protected].
FAQs
Q: What standards apply to medical electronic devices?
A: IEC-60601, IEC-61000, and ISO 14971 are the primary standards.
Q: What is the biggest mistake in design?
A: Ignoring regulations early, leading to redesign.
Q: What is 510(k)?
A: A submission proving equivalence to an existing device.
Q: Why is patient isolation important?
A: It prevents electrical harm and ensures safety compliance.
Ask A Technical Question
Stay up-to-date on Chemtronics news, products, videos & more.
Related Products




